Releases: FelixKrueger/Bismark
Releases · FelixKrueger/Bismark
v0.16.0
Bismark
- File endings
.fastq | .fq | .fastq.gz | .fq.gzare now removed from the output file (unless they were specified with--basename) in a bid to reduce the length of the already long file names. - Enabled the new option
--dovetail(which will be turned on by default for--pbatlibraries) which will now allow dovetailing reads to be reported. For a more in-depth description see #14. - Changed the behaviour of corner cases to where several non-directional alignments could have existed for the very same position but to different strands so that now the best alignment trumps the weaker one. As an example: If you relaxed the alignment criteria of a given alignment to allow ~60 mismatches for PE alignment we did find an alignment to the OT strand with a combined AS of -324, but there also was an alignment to the CTOB strand with and AS of 0 (perfect alignment). The CTOB now trumps the OT alignment, and the methylation information information is now reported for the bottom strand. Credits go to Sylvain Foret (ANU, Canberra) for bringing this to our attention!
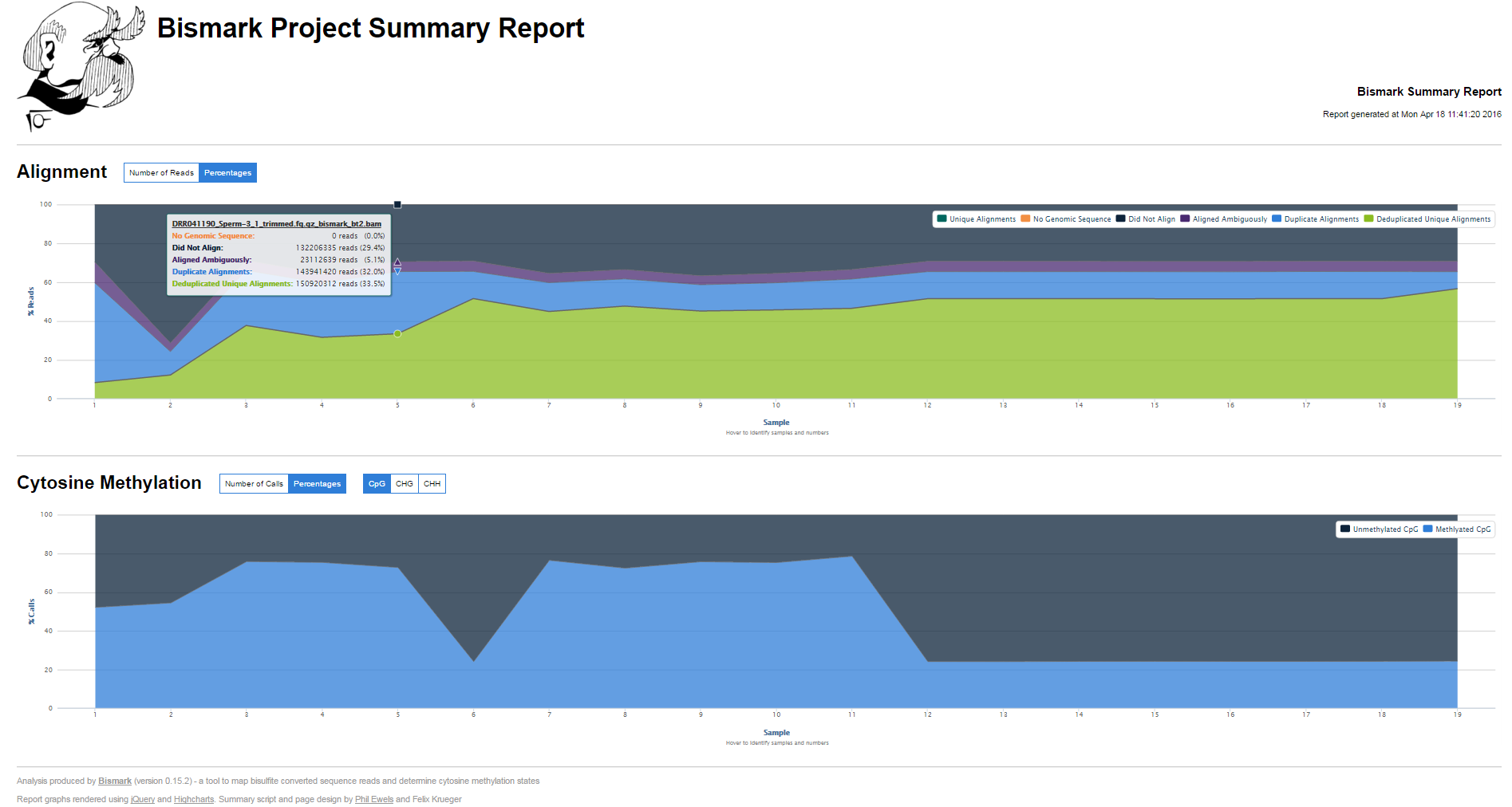
New module: bismark2summary
bismark2summaryaccepts Bismark BAM files as input. It will then try to identify Bismark reports, and optionally deduplication reports or methylation extractor (splitting) reports automatically based the BAM file basename. It produces a tab delimited overview table (.txt) as well as a graphical HTML report.
Examples can be found at http://www.bioinformatics.babraham.ac.uk/projects/bismark/bismark_summary_report.html and http://www.bioinformatics.babraham.ac.uk/projects/bismark/bismark_summary_report.txt. Thanks to @ewels for help with the Java Script part!
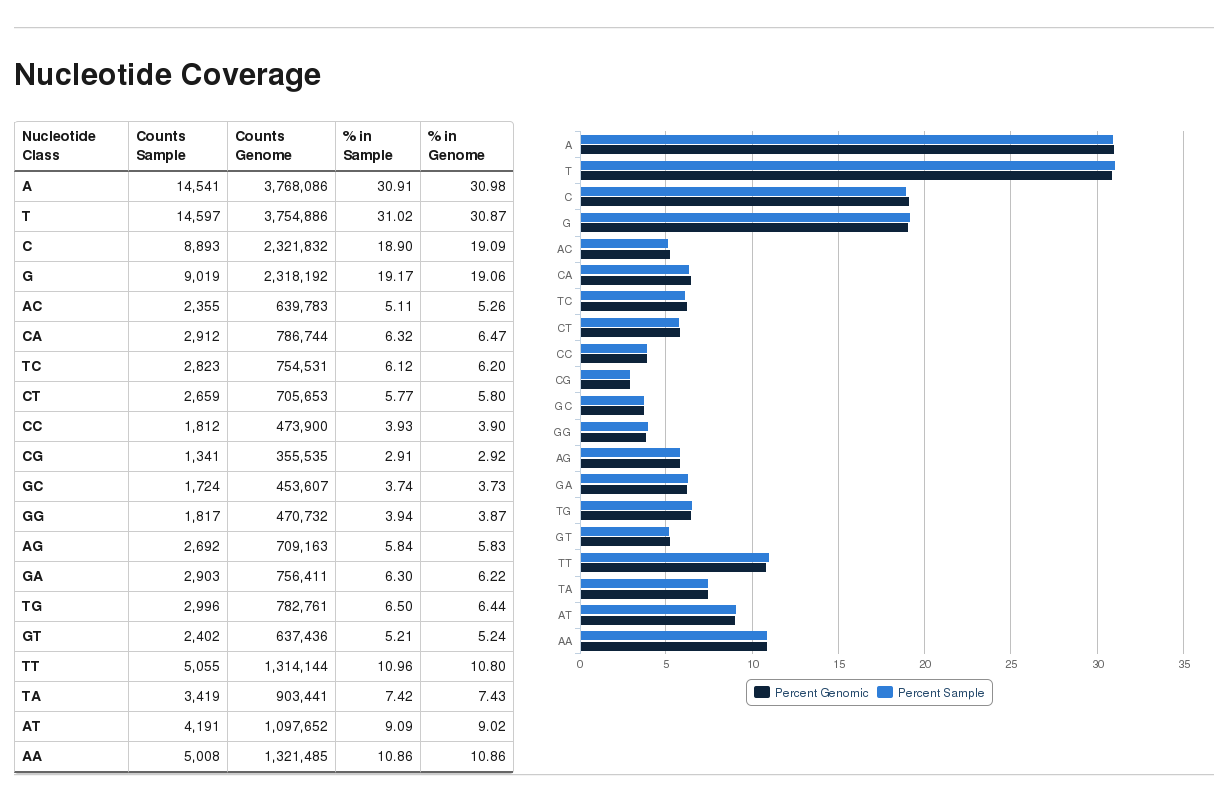
New module: bam2nuc
- The new Bismark module
bam2nuccalculcates the average mono- and di-nucleotide coverage of libraries and compares this to the genomic average composition.bam2nuccan be called straight from within Bismark (option--nucleotide_coverage) or run stand-alone.bam2nuccreates a...nucleotide_stats.txtfile that is also automatically detected bybismark2reportand incorporated into the HTML report.
bismark2_sitrep.tpl
- Removed an extra function call in
bismark_sitrep.tplso that the M-bias 2 plot is drawn once the M-bias 1 plot has finished drawing (parallel processing could with certain browsers and data may have resulted in a white spaceholder only).
methylation extractor
- Altering the file path handling of
coverage2cytosineandbismark2bedGraphalso required some changes in the methylation extractor.
bismark2bedGraph
- Input file path handling has been completely reworked. The output file which can be specified as
-o output.bedGraphnow has to be a single file name and mustn't contain any path information. A particular output folder may be specified with-dir /any/path/. - Addressing the file path handling issue also fixed a similar issue with the option
--remove_spaceswhen-ohad been specified.
coverage2cytosine
- Changed
zcatforgunzip -cwhen reading a gzipped coverage file. This should avoid some Mac platforms crashing because zcat invariably requires a file to end in the .Z (which it doesn't...) - Changed the way in which the coverage input file is handed over from the
methylation_extractor
tocoverage2cytosine(previously the path information might have been part of the file name, but
instead it will now be only part of the-dir output_directoryoption.
v0.15.0
Bismark
- Added option
--se/--single_end <list>. This sets single-end mapping mode explicitly giving a
list of file names as<list>. The filenames may be provided as a comma,or colon:-separated
list. - Added option
--genome_folder <path/to/genome>as alternative to supplying the genome as the
first argument. - Added an option
--rg_tagto print an@RG header lineas well as andRG:Z:tag to each read.
The ID and SAMPLE fields default to 'SAMPLE', but can be specified manually with--rg_idor
--rg_sample. - Added new option
--ambig_bamfor Bowtie2-mode only, which writes out a single alignment for
sequences with multiple alignments to a special file ending in.ambiguous.bam. The alignments
are in Bowtie2 format and do not any contain Bismark specific entries such as the methylation
call etc. These ambiguous BAM files are intended to be used as coverage estimators for variant
callers. Works for single-end and paired-end alignments in single or multi-core mode. - Added the new options
--cramand--cram_refto Bismark for both paired- and single-end alignments
in single or multi-core mode. This option requires Samtools version 1.2 or higher. A genome
FastA reference may be supplied as a single file with the option--cram_ref; if this is not
specified the file is derived from the reference FastA file(s) used for the Bismark run, and written
to the fileBismark_genome_CRAM_reference.mfainto the output directory.
deduplicate_bismark
- Added better handling of cases when the input file was empty (died for percentage calculation
instead of calling it N/A) - Added a note mentioning that Read1 and Read2 of paired-end files are expected to follow each
other in two consecutive lines and possibly require name-sorting prior to deduplication. Also
added a check that reads the first 100000 lines to see if the file appears to have been sorted
and bail out if this is true.
methylation extractor
- Added support for
CRAMfiles (this option requires Samtools version 1.2 or higher)
bismark2bedGraph
- Changed the way
gzipcompressed input files are handled when using theUNIX sortcommand (i.e. with
--scaffolds/--gazillionor without--ample_memory
coverage2cytosine
- Added option
--gzipto compress output files. This currently only works for the default CpG_report
and CX_report output files (and thus not with the option--gcor--split_files. The option--gzip
is now also passed on from thebismark_methylation_extractor. - Added a check to bail if no information was found in the coverage file, e.g. if a wrong file path for a .cov.gz file had been specified
bismark_genome_preparation
- Added process handling to the child processes.
Bismark v0.14.5
20-08-2015: 0.14.5 released - minor fix
- deduplicate_bismark: Changed all instances of literal calls of
samtoolscalls to$samtools_path